参考中国报告网发布《2016-2022年中国化学药品制剂市场深度调查及十三五未来前景预测报告》
近年来,已在海外规范市场上市的仿制药在审评、一致性评价和招标采购分组等方面享受到了诸多优惠政策。从审评方面来看,2016 年初 CFDA 发布了《关于解决药品注册申请积压实行优先审评审批的意见》,其中要求对“在中国境内用同一生产线生产并在美国、欧盟药品审批机构同步申请上市且通过了其现场检查的药品注册申请”实施优先申请审批,自该项政策发布 1 年多以来,共有 15 个品种,涉及 8 个厂家的注册申请受益于该项政策。在被列入优先审评序列之后,审评的速度有望大大提高,加快了其在国内上市的步伐。
在仿制药的上市程序方面,如果原研药已在国内上市,一般首先需要进行 BE试验,然后再报生产。在 CFDA 于 5 月 11 日发布的《关于鼓励药品医疗器械创新改革临床试验管理的相关政策》(征求意见稿)意见》之中提出:申请人在欧洲药品管理局、美国和日本获准上市仿制药的生物等效性试验数据,符合中国药品注册相关要求的,经现场检查后可用于在中国申报仿制药注册。
在 6 月 9 日发布的《关于仿制药质量和疗效一致性评价工作有关事项的公告(征求意见稿)》也提出:在欧盟、美国或日本上市但未在中国境内上市的,经临床研究证实无种族差异的,可使用境外上市申报的生物等效性研究、药学研究数据等技术资料向食品药品监管总局(药品审评中心)提出上市申请。该项政策的推出使得已实现海外上市的药品在国内上市时免于重复进行 BE 试验,可以以海外试验数据为基础申请在国内注册,上市流程被进一步简化。
再加上已经推出的优先审评政策,已在海外规范市场上市的仿制药回归国内的步伐将大大加快。 对于在国内和海外规范市场(美国、欧盟和日本)均以上市的仿制药,在《关于仿制药质量和疗效一致性评价工作有关事项的公告(征求意见稿)》中按照生产线和处方工艺是否变更进行了区分,在其通过一致性评价方面制定了不同的政策。
对于生产线和处方工艺未变更的经过审评后即可视为通过一致性评价;对于发生变更的,则要求进行处方工艺变更,再视作通过一致性评价,程序上虽然有所增加,但是相比于其他普通产品相比一方面免于进行药学研究、BE 试验等事项,节省了时间和费用,另一方由于在海外上市时对于生物等效性、药学研究等方面的要求较为严格,这些药品通过审评的概率也较大。

在《关于仿制药质量和疗效一致性评价工作有关事项的公告(征求意见稿)》中提出:对正在审评中的按照原化学药品注册分类受理的仿制药注册申请,申请人可向食品药品监管总局提出按与原研药质量和疗效一致的标准审评的申请。审评通过的,视为通过一致性评价。对于海外已上市申请在国内上市的仿制药,可以利用这一规则可以同步实现通过一致性评价和国内上市,实现弯道超车。
通过对于政策的梳理,可以看出在制剂出口领域,海外已上市产品转报国内和已实现国内外同步上市的产品均能够迎来利好,最终能够在产品上市速度和一致性评价进度方面占得先机。对于国内外均以上市的产品,由于其销售渠道、营销经验已经较为成熟,通过一致性评价后可以迅速兑现预期。
对于海外已上市转报国内的产品,如果原有的销售渠道不够健全,营销经验不够充足,企业仍然需要充分利用率先通过一致性评价的时间优势,加强营销力量,最终实现替代进口产品和市场份额的扩大。在制剂出口领域,我们建议关注华海药业(制剂出口龙头,多个品种获得美国 ANDA 批准文号)和普利制药(深耕注射剂出口领域,多个海外上市品种获得优先审评资格)。

近年来,已在海外规范市场上市的仿制药在审评、一致性评价和招标采购分组等方面享受到了诸多优惠政策。从审评方面来看,2016 年初 CFDA 发布了《关于解决药品注册申请积压实行优先审评审批的意见》,其中要求对“在中国境内用同一生产线生产并在美国、欧盟药品审批机构同步申请上市且通过了其现场检查的药品注册申请”实施优先申请审批,自该项政策发布 1 年多以来,共有 15 个品种,涉及 8 个厂家的注册申请受益于该项政策。在被列入优先审评序列之后,审评的速度有望大大提高,加快了其在国内上市的步伐。
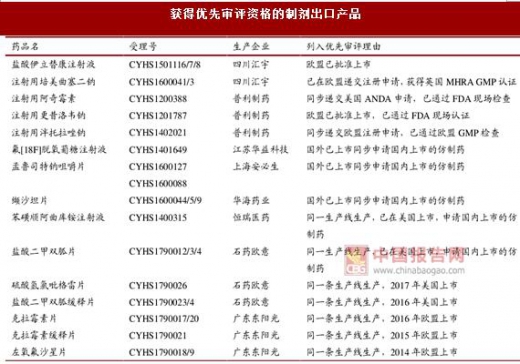
获得优先审评资格的制剂出口产品

资料来源:中国报告网整理
在仿制药的上市程序方面,如果原研药已在国内上市,一般首先需要进行 BE试验,然后再报生产。在 CFDA 于 5 月 11 日发布的《关于鼓励药品医疗器械创新改革临床试验管理的相关政策》(征求意见稿)意见》之中提出:申请人在欧洲药品管理局、美国和日本获准上市仿制药的生物等效性试验数据,符合中国药品注册相关要求的,经现场检查后可用于在中国申报仿制药注册。
在 6 月 9 日发布的《关于仿制药质量和疗效一致性评价工作有关事项的公告(征求意见稿)》也提出:在欧盟、美国或日本上市但未在中国境内上市的,经临床研究证实无种族差异的,可使用境外上市申报的生物等效性研究、药学研究数据等技术资料向食品药品监管总局(药品审评中心)提出上市申请。该项政策的推出使得已实现海外上市的药品在国内上市时免于重复进行 BE 试验,可以以海外试验数据为基础申请在国内注册,上市流程被进一步简化。
再加上已经推出的优先审评政策,已在海外规范市场上市的仿制药回归国内的步伐将大大加快。 对于在国内和海外规范市场(美国、欧盟和日本)均以上市的仿制药,在《关于仿制药质量和疗效一致性评价工作有关事项的公告(征求意见稿)》中按照生产线和处方工艺是否变更进行了区分,在其通过一致性评价方面制定了不同的政策。
对于生产线和处方工艺未变更的经过审评后即可视为通过一致性评价;对于发生变更的,则要求进行处方工艺变更,再视作通过一致性评价,程序上虽然有所增加,但是相比于其他普通产品相比一方面免于进行药学研究、BE 试验等事项,节省了时间和费用,另一方由于在海外上市时对于生物等效性、药学研究等方面的要求较为严格,这些药品通过审评的概率也较大。
制剂出口产品通过一致性评价的相关政策

资料来源:中国报告网整理
在《关于仿制药质量和疗效一致性评价工作有关事项的公告(征求意见稿)》中提出:对正在审评中的按照原化学药品注册分类受理的仿制药注册申请,申请人可向食品药品监管总局提出按与原研药质量和疗效一致的标准审评的申请。审评通过的,视为通过一致性评价。对于海外已上市申请在国内上市的仿制药,可以利用这一规则可以同步实现通过一致性评价和国内上市,实现弯道超车。
通过对于政策的梳理,可以看出在制剂出口领域,海外已上市产品转报国内和已实现国内外同步上市的产品均能够迎来利好,最终能够在产品上市速度和一致性评价进度方面占得先机。对于国内外均以上市的产品,由于其销售渠道、营销经验已经较为成熟,通过一致性评价后可以迅速兑现预期。
对于海外已上市转报国内的产品,如果原有的销售渠道不够健全,营销经验不够充足,企业仍然需要充分利用率先通过一致性评价的时间优势,加强营销力量,最终实现替代进口产品和市场份额的扩大。在制剂出口领域,我们建议关注华海药业(制剂出口龙头,多个品种获得美国 ANDA 批准文号)和普利制药(深耕注射剂出口领域,多个海外上市品种获得优先审评资格)。
制剂出口生产企业受益方向

资料来源:互联网
资料来源:中国报告网整理,转载请注明出处(GQ)
更多好文每日分享,欢迎关注公众号

【版权提示】观研报告网倡导尊重与保护知识产权。未经许可,任何人不得复制、转载、或以其他方式使用本网站的内容。如发现本站文章存在版权问题,烦请提供版权疑问、身份证明、版权证明、联系方式等发邮件至kf@chinabaogao.com,我们将及时沟通与处理。