医药产业研发现状
新药研发作为医药产业链的上游,其发展状况直接影响到我国医药产业未来的生存和发展。随着国家鼓励创新政策的出台,以及国家研究中心和科研院所的技术支撑,我国一类新药的申报数量有所增加,相应的专利申报数量也有较大幅度的提高,医药行业的整体创新能力有所增强。同时随着社会分工的进一步专业化和细化,一批以CRO为主业的本土企业逐步成长起来并在A股上市(CRO企业即以医药研发为主业的企业,服务范围涵盖医药研发流程的各环节,包括临床前CRO和临床试验CRO,可根据客户需求提供特定环节的定制化服务,上市公司泰格医药为典型CRO企业)。
不过,当前国内医药研发行业仍存在诸多问题:研发主体错位,制药水平先进的国家以企业为新药研发的主体,而国内主要由专业院校和研究院来承担;知识产权保护意识有待加强,中药老字号商标及专利在海外遭抢注,使得国内企业蒙受损失;研发投入严重不足,这是国内医药研发面临的最大问题。一方面专业院校与研究院研发资金来源政府,资金规模有限,另一方面医药制造行业集中低,企业规模普遍偏小,亦难以有效支撑高水平的研发。国际大型制药公司一般将利润的10~20%用于研发,2015年国内医药制造行业技术投入比率(企业本年科技支出与本年营业收入的比率)仅为3.3%,2016年医药制造上市企业的研发支出/营业收入亦处于较低水平(中位数为4.28%)。
医药作为涉及所有人身体健康甚至生命安全的消费品,在研发、制造及流通环节均受到严格的监管。在医药研发的临床前研究及临床试验过程中,监管部门均制定了相应的质量管理规范,同时在关键节点设立审评制度,从源头来确保药品的疗效及安全性。
参考中国报告网发布《2017-2022年中国医药行业市场发展现状及未来前景分析报告》
在药品研发过程中最为重要的质量管理规范为《药物非临床研究质量管理规范》和《药物临床试验质量管理规范》,即GLP和GCP。GLP和GCP对临床前研究及临床试验均制定了严格的要求,明确了相应的权责,从流程上来保障药品研发的质量。此外CFDA针对研发过程中的各环节制定了相应的技术指导原则。
药品监管部门除了制定系列质量管理规范和技术指导原则,还在医药研发的过程中设置了两个重要的审评环节,一个是药品临床试验申请,另外一个是药品上市申请。根据《药品注册管理办法》的规定研发新药必须按照国家药品监督管理部门的规定如实报送研制方法、质量指标、药理及毒理试验结果等有关资料和样品,经国家药品监督管理部门审核批准后,方可进行临床试验,如下图所示。完成临床试验后并通过审评的新药,由国家药品监督管理部门批准,颁发新药证书,至此药品研发成功,申报流程与临床试验申报流程基本一致。

药品研发两个关键节点的审评制度有利于在源头上保障药品质量,但目前药品注册审评积压较为严重,审评阶段耗费时间较长(时间成本偏高),使得审批耗时成为企业在进行药品研发时需要重点考虑的因素。根据Insight-ChinaPharmaData数据库统计,2011~2014年获得批准的1.1类新药中:申报临床试验的平均审评时间为14个月,申报生产的平均审评时间为29个月(以获得生产批件为主,拥有新药证书的企业,需要通过生产审评后获得生产批件,方可进行药品的生产)。造成审评效率较低(药品注册审评积压)主要有两方面原因:一方面审评人员相对偏少,人均审评工作量偏大,另一方审评机制有待进一步完善,原有审评机制导致很多企业提前进行立项和申报,造成部分的虚假申报、仿制药重复申请,进一步加重了审评积压的问题。目前随着优先审批政策、仿制药一致性评价以及关于《开展药物临床试验数据自查核查工作的公告》的颁布,有望清理注册挤压、压缩审评环节的时间占用。
新药研发作为医药产业链的上游,其发展状况直接影响到我国医药产业未来的生存和发展。随着国家鼓励创新政策的出台,以及国家研究中心和科研院所的技术支撑,我国一类新药的申报数量有所增加,相应的专利申报数量也有较大幅度的提高,医药行业的整体创新能力有所增强。同时随着社会分工的进一步专业化和细化,一批以CRO为主业的本土企业逐步成长起来并在A股上市(CRO企业即以医药研发为主业的企业,服务范围涵盖医药研发流程的各环节,包括临床前CRO和临床试验CRO,可根据客户需求提供特定环节的定制化服务,上市公司泰格医药为典型CRO企业)。
不过,当前国内医药研发行业仍存在诸多问题:研发主体错位,制药水平先进的国家以企业为新药研发的主体,而国内主要由专业院校和研究院来承担;知识产权保护意识有待加强,中药老字号商标及专利在海外遭抢注,使得国内企业蒙受损失;研发投入严重不足,这是国内医药研发面临的最大问题。一方面专业院校与研究院研发资金来源政府,资金规模有限,另一方面医药制造行业集中低,企业规模普遍偏小,亦难以有效支撑高水平的研发。国际大型制药公司一般将利润的10~20%用于研发,2015年国内医药制造行业技术投入比率(企业本年科技支出与本年营业收入的比率)仅为3.3%,2016年医药制造上市企业的研发支出/营业收入亦处于较低水平(中位数为4.28%)。
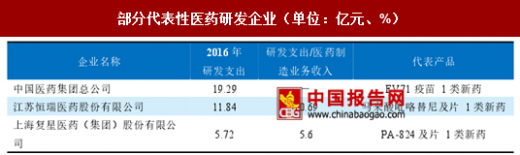
部分代表性医药研发企业(单位:亿元、%)

资料来源:中国报告网整理
研发环节相关制度与政策医药作为涉及所有人身体健康甚至生命安全的消费品,在研发、制造及流通环节均受到严格的监管。在医药研发的临床前研究及临床试验过程中,监管部门均制定了相应的质量管理规范,同时在关键节点设立审评制度,从源头来确保药品的疗效及安全性。
参考中国报告网发布《2017-2022年中国医药行业市场发展现状及未来前景分析报告》
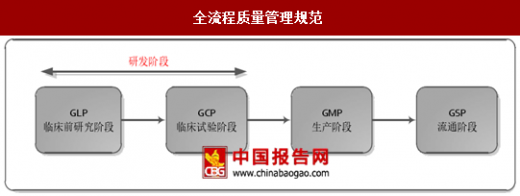
在药品研发过程中最为重要的质量管理规范为《药物非临床研究质量管理规范》和《药物临床试验质量管理规范》,即GLP和GCP。GLP和GCP对临床前研究及临床试验均制定了严格的要求,明确了相应的权责,从流程上来保障药品研发的质量。此外CFDA针对研发过程中的各环节制定了相应的技术指导原则。
全流程质量管理规范

资料来源:中国报告网整理
药品监管部门除了制定系列质量管理规范和技术指导原则,还在医药研发的过程中设置了两个重要的审评环节,一个是药品临床试验申请,另外一个是药品上市申请。根据《药品注册管理办法》的规定研发新药必须按照国家药品监督管理部门的规定如实报送研制方法、质量指标、药理及毒理试验结果等有关资料和样品,经国家药品监督管理部门审核批准后,方可进行临床试验,如下图所示。完成临床试验后并通过审评的新药,由国家药品监督管理部门批准,颁发新药证书,至此药品研发成功,申报流程与临床试验申报流程基本一致。
新药临床试验申报流程及监管部门

资料来源:中国报告网整理
药品研发两个关键节点的审评制度有利于在源头上保障药品质量,但目前药品注册审评积压较为严重,审评阶段耗费时间较长(时间成本偏高),使得审批耗时成为企业在进行药品研发时需要重点考虑的因素。根据Insight-ChinaPharmaData数据库统计,2011~2014年获得批准的1.1类新药中:申报临床试验的平均审评时间为14个月,申报生产的平均审评时间为29个月(以获得生产批件为主,拥有新药证书的企业,需要通过生产审评后获得生产批件,方可进行药品的生产)。造成审评效率较低(药品注册审评积压)主要有两方面原因:一方面审评人员相对偏少,人均审评工作量偏大,另一方审评机制有待进一步完善,原有审评机制导致很多企业提前进行立项和申报,造成部分的虚假申报、仿制药重复申请,进一步加重了审评积压的问题。目前随着优先审批政策、仿制药一致性评价以及关于《开展药物临床试验数据自查核查工作的公告》的颁布,有望清理注册挤压、压缩审评环节的时间占用。
资料来源:中国报告网整理,转载请注明出处(ZQ)
更多好文每日分享,欢迎关注公众号

【版权提示】观研报告网倡导尊重与保护知识产权。未经许可,任何人不得复制、转载、或以其他方式使用本网站的内容。如发现本站文章存在版权问题,烦请提供版权疑问、身份证明、版权证明、联系方式等发邮件至kf@chinabaogao.com,我们将及时沟通与处理。