药用辅料行业行业主管部门
药用辅料是药品生产的重要组成部分,我国一直以来将药用辅料参照药品管理,药用辅料生产企业的日常经营行为必须遵循国家医药行业管理的相关政策法规。
我国医药行业主管部门是国家食品药品监督管理总局,负责对全国医药市场进行监督管理。国家发展和改革委员会、国家卫生和计划生育委员会、国家工业和信息化部等部门与国家食品药品监督管理总局共同对医药行业进行监督与管理,地方各级食品药品监督管理部门负责本行政区域内的药品监督管理工作。
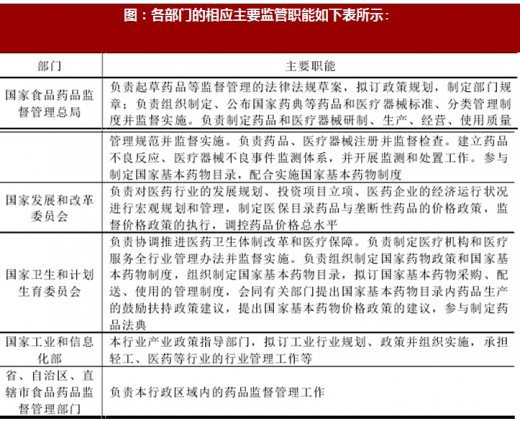
图:各部门的相应主要监管职能如下表所示:
药用辅料作为药品生产的重要组成部分,《中华人民共和国药品管理法》规定“生产药品所需的原料、辅料,必须符合药用要求”。我国药用辅料参照药品管理,药用辅料生产企业的日常经营行为必须遵循国家医药行业管理的相关政策法规。
随着药品监管工作的不断深入,行业主管部门对加强药用辅料规范管理的要求不断提高,推动我国药用辅料行业的法规体系和质量标准不断完善。目前行业适用的法律法规主要包括:《中华人民共和国药品管理法》及其实施条例、《中华人民共和国药典》02015年版)、《药品生产监督管理办法》、《药品生产质量管理规范(2010年修订)》及《药用辅料生产质量管理规范》等,具体如下:
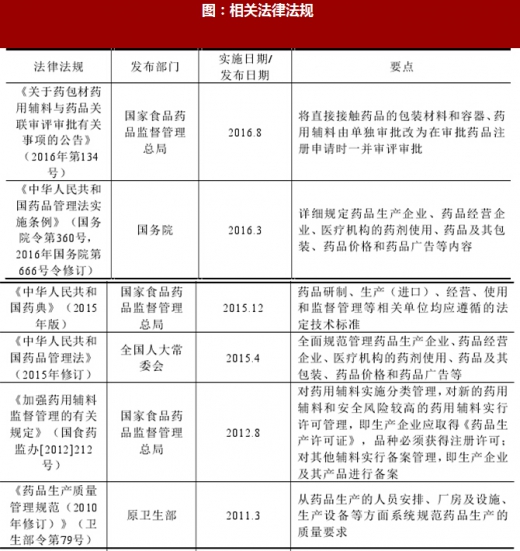
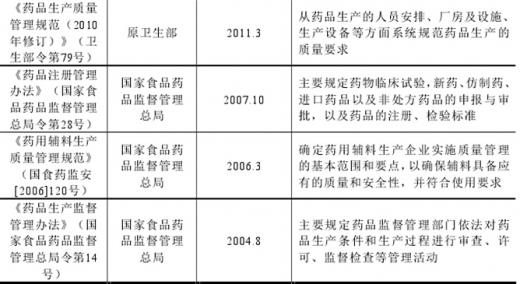
图:相关法律法规
行业政策
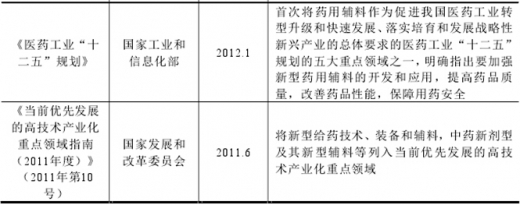
图:药用辅料相关的主要行业政策
①药用辅料分类管理制度及药用辅料关联评审制度
《加强药用辅料监督管理的有关规定》(国食药监办[2012]212号)规定,对药用辅料实施分类管理,对新的药用辅料和安全风险较高的药用辅料实行许可管理,即生产企业应取得《药品生产许可证》,品种必须获得注册许可;对其他辅料实行备案管理,即生产企业及其产品进行备案。对实施许可管理的药用辅料,生产企业应按要求提交相关资料。对实施备案管理的药用辅料,由生产企业提交相关资料,报所在地省级食品药品监督管理部门备案。《关于印发药用辅料注册申报资料要求的函》(食药监注函【2005]61号)规定,新药用辅料和进口药用辅料由国家局审批,己有国家标准药用辅料由省局审批。
参考观研天下发布《2017-2022年中国药用食品行业市场发展现状及十三五市场竞争态势报告》
2016年8月,国家食品药品监督管理总局发布《总局关于药包材药用辅料与药品关联审评审批有关事项的公告》02016年第134号),明确“将直接接触药品的包装材料和容器(以下简称药包材)、药用辅料由单独审批改为在审批药品注册申请时一并审评审批”。药用辅料应按程序与药品注册申请关联申报和审评审批。各级食品药品监督管理部门不再单独受理药用辅料注册申请,不再单独核发相关注册批准证明文件。己批准的药包材、药用辅料,其批准证明文件在有效期内继续有效。有效期届满后,可继续在原药品中使用。如用于其他药品的药物临床试验或生产申请时,需进行评审。
②药品生产许可制度
根据《中华人民共和国药品管理法》的规定,在我国开办药品生产企业,须经企业所在地省、自治区、直辖市人民政府药品监督管理部门批准并发给《药品生产许可证》,《药品生产许可证》应当标明有效期和生产范围,到期重新审查发证。无《药品生产许可证》的,不得生产药品。经省、自治区、直辖市人民政府药品监督管理部门批准,药品生产企业可以接受委托生产药品。在我国境内生产实施许可管理的药用辅料,需取得《药品生产许可证》。
③国家药品标准制度
国家药品标准是指国家为保证药品质量所制定的质量指标、检验方法以及生产工艺等技术要求,包括国家食品药品监督管理总局颁布的《中华人民共和国药典》、药品注册标准和其他药品标准。《中国药典》是国家药品标准体系的核心内容,《中国药典》2015版在品种收载、标准体系的系统完善、质控水平的整体提升方面进一步完善了药品及辅料标准体系。药用辅料独立成一卷,构成《中国药典》四部的主要内容;药用辅料品种收载数量显著增加,增至270种;同时,载入《中国药典》的注射用药用辅料从2010年版的1个增加到2015年版的13个,新增12个之多。
④药用辅料生产质量管理制度
2006年3月,国家食品药品监督管理总局颁布了《药用辅料生产质量管理规范》,从机构、人员和职责、厂房和设施、设备、物料、卫生、验证、文件、生产管理、质量保证和质量控制、销售以及自检和改进等方面较为全面、系统地规定了药用辅料生产质量管理规范要求。2012年8月,国家食品药品监督管理总局颁布了《加强药用辅料监督管理的有关规定》(国食药监办【2012]212号),进一步加强药用辅料生产、使用的监管。
资料来源:观研天下整理,转载请注明出处(ZTT)
更多好文每日分享,欢迎关注公众号

【版权提示】观研报告网倡导尊重与保护知识产权。未经许可,任何人不得复制、转载、或以其他方式使用本网站的内容。如发现本站文章存在版权问题,烦请提供版权疑问、身份证明、版权证明、联系方式等发邮件至kf@chinabaogao.com,我们将及时沟通与处理。