生物类似药尤其是以抗体药为主的新一代生物类似药总体还处于初期阶段,各国政策正在以不同的速度发生改变以适应生物类似药的发展,比如欧洲是生物类似药发展最早最快的地区,美国近年来也加快了步伐,印度作为一个不规范市场,制度还需要进一步的完善,中国也在不断的加强制度改革以适应国内强大的研发上市需求。我们认为政策的变化对于生物类似药的投资有很强的影响,所以在此总结各个主要国家和地区在生物类似药上的政策变革,以期对生物类似药的投资有所帮助。
一、美国:近几年步伐加快,生物类似药监管体系已基本完善
1984 年,美国通过“药品价格竞争及专利补偿法案”(Hatch—Waxman 法案),仿制药的概念就始于此,此法案为仿制药(主要是针对化学药)进入市场提供简化申请程序。
2009 年,美国国会通过“生物制品价格竞争和创新法案” (BPCIA),奠定了生物类似药批准途径的法律基础,并设立了具体的申请途径 351(K)途径。
2010 年,颁布了《患者保护与平价医疗法案》,制定了生物仿制药进入市场的简化申请途径,即“生物仿制药途径”,通过该途径递交的生物类似药申请称为 351(K)申请。明确了生物类似药参比产品的监管排他期,新药获批 4 年内,不得向 FDA 申请生物类似药,12 年内,不得上市类似药。这意味着生物创新药享有 12 年市场独占权,在此期间没有类似药上市与之竞争。
2012 年,FDA 公布的指导文件草案中描述了生物类似药在美国批准的框架。
2014 年,出台了《阐明与参照药的生物相似性的临床药理数据指导原则》。
2012-2015 年,FDA 出台了 7 部指南来执行上述立法。
2017 年 6 月 12 日,美国最高法院裁决安进和山德士之间的“专利之舞”案件,判定生物类似药在获得 FDA 批准后不必再等待 6 个月才能上市,这一事件对于生物类似药的上市具有重大意义。

二、欧盟:发展最早、最完善,生物类似药监管体系已成全球领头羊
欧盟的医保体系相对最为完善,欧盟药品管理局(EMA)建立了完善的政策法规体系,而且不断的进行完善升级,在全球生物类似药的开发和监管上起着示范和引领作用。
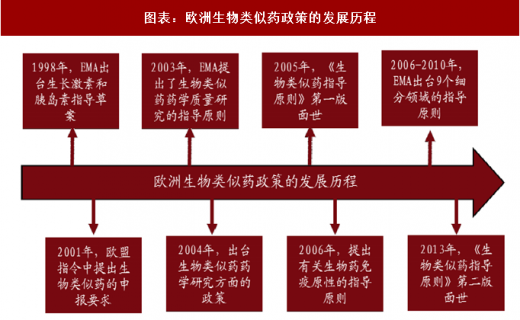
1998 年,EMA 出台生长激素和胰岛素指导草案。
2001 年,欧盟指令 2001/83/EC 中提出了生物类似药的申报要求。
2003 年,EMA 出台基本的法律条文,率先进行生物类似药指导原则的制定,提出了生物类似药药学质量研究的指导原则。
2004 年,进一步出台了有关生物类似药药学研究方面的政策。
2005 年,EMA 正式提出了有关生物类似药的一系列指南,《生物类似药指导原则》第一版面世。
2006 年,提出了有关生物药免疫原性的指导原则,为生物类似药的免疫原性研究提供重要的指导作用。
2006-2010 年,EMA 出台了 9 个细分领域的指导原则。主要是针对胰岛素、生长激素、粒细胞集落刺激因子、促红细胞生成素、促卵泡素、小分子肝素、单抗、重组人干扰素 α 和重组人干扰素 β 领域颁布的具体指导原则。
参考观研天下发布《2018年中国医药零售行业分析报告-市场运营态势与发展趋势研究》
2013 年,《生物类似药指导原则》第二版面世。
以上指导原则重点强调的是生物类似药在质量、安全性和有效性上与原研产品一致。2006 年,EMA 批准了第一个真正意义上的生物类似药,即生长激素生物类似药 Omnitrope。欧洲也自此开启了生物类似药的篇章。欧盟生物类似药的法律法规体系最健全,发展也最早最快。
截止到 2017 年 5 月,EMA 批准的生物类似药超过了 30 款。其中 2017 年有 7 款生物类似药获批,3 款是美罗华的类似药,2 款修美乐的类似药,1 款恩利的类似药,1 款来得时的类似药。
三、印度:宽松制度下,生物药类似药疯狂发展
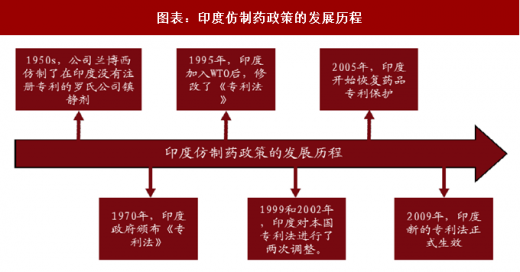
1950s,印度还在沿用英国统治时期的产品专利法,制药受到严格控制,且印度研发能力较弱,药靠进口,费用高,民众消费不起。
1970 年,印度政府颁布《专利法》,规定只保护制药工艺,不保护药品成分,为仿制药开启绿灯。只要工艺流程不同,就可随意仿制生产任意一种药品分子,自此仿制药在印度开始疯狂发展。
1995 年,印度加入 WTO 后,修改了《专利法》。
1999 和 2002 年,在欧美等国的强烈要求和 WTO 监督下,印度对本国专利法进行了两次调整。
2005 年,印度开始恢复药品专利保护,对 1995 年以后发明或改造的药物提供专利保护,对原有药物混合或衍生药物无效。
2009 年,印度新的专利法正式生效,专利政策收紧,但相对于其他国家仍然很宽松。近些年来印度的制药公司也一直受到跨国药企的专利诉讼,但通常以跨国药企败诉收场。 一些跨国药企退而求其次,选择和印度的生物制药公司合作,比如辉瑞与博枞公司合作生产的胰岛素在印度上市,吉列德将 Sovaldi 免费授权给印度公司生产,允许他们将药销售给贫穷国家。
由于印度政策宽松,对仿制药的大力支持,使得印度的仿制速度很快,研发能力在仿制药不断做强后也随之增强,靠仿制药做大的药企也开始注重创新药的研发并积极和国际制药巨头合作占领市场。在印度已上市的生物类似药已超过 60 种,如此多的生物类似药,使得印度的生物药价格极低。
四、中国:生物类似药正在起步,政策正逐步完善
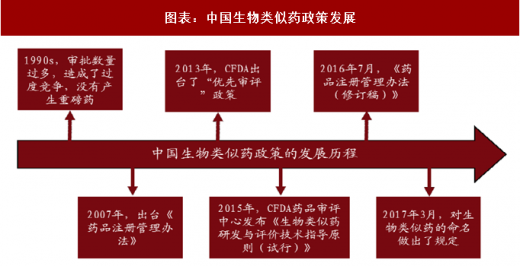
1990s,中国的生物制药由于没有严格的审批制度,审批数量过多,造成了过度竞争,没有产生重磅药。
2007 年,出台《药品注册管理办法》,规定所有生物制品均按照新药来申报,限制了同一药品的企业数量。
2013 年,国家食品药品监督管理总局(CFDA)出台了“优先审评”政策,这个“优先审评”只针对于仿制药。
2015 年 2 月,CFDA 药品审评中心(CDE)发布《生物类似药研发与评价技术指导原则(试行)》,确定了生物类似药的监管框架,界定了生物类似药及参比药的定义,规定了技术审查的基本原则、比对标准和适应症外推的条件。
2016 年 7 月,《药品注册管理办法(修订稿)》,进一步规范了生物类似药的概念,严格了生物类似药的审批标准。2017 年 3 月,对生物类似药的命名做出了规定,如果是按照类似药的标准进行研究开发和临床试验的类似药可以和原研药用相同的名字,可考虑适应症外推。
CDE 允许企业走新药或生物类似药两条途径展开临床试验。
走新药临床开发途径可避免在 III 期临床购买原研药的巨额花费,同时也可避免无法达到与原研药相似的标准;
按照类似药途径申请的生物类似药可能会获得适应症外推的利好,即一款药如果被证明在一个病症上和原研相似,原研药的其他适应症也可直接适用于这款药。

【版权提示】观研报告网倡导尊重与保护知识产权。未经许可,任何人不得复制、转载、或以其他方式使用本网站的内容。如发现本站文章存在版权问题,烦请提供版权疑问、身份证明、版权证明、联系方式等发邮件至kf@chinabaogao.com,我们将及时沟通与处理。